
What is a Combination Product?
In a world in which we are continuously challenged to operate more efficiently and immersed in rapid technological advances, it should come as no surprise that one of today’s fastest growing medical product segments are combination products. To be considered a combination product by FDA, there must be two or more regulated components, (i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic) that are physically, chemically, or otherwise combined or mixed and produced as a single entity, or two separate products that are packaged together and per proposed labeling are intended for use together to achieve a desired effect.[1] Combination products should not be confused with Fixed-Combination Drugs (FCDs), which are drug-drug combinations.
For the purpose of this article, we will focus on the drug/device segment. We do so because we are so often called upon to address drug delivery devices. Not surprisingly, “High incidence rates of chronic pain causing diseases, prostate cancer, diabetic retinopathy, cardiovascular diseases, colorectal cancer, asthma, obesity along with rapidly aging global population also form the key drivers for the global drug device combination products market.”[2] However, much of the information herein may be extrapolated to other facets of combination products.
Who Reviews a Combination Product?
Combination products are assigned to Centers with primary jurisdiction for premarket reviews and regulation. The determination is based on “primary mode of action” (PMOA) of the combination product.[3]
Which FDA Center Will Review my Product?
As you might imagine, combination products face unique regulatory hurdles regarding review, pathway, post-market regulation, etc. There is not always a clear-cut pathway. However, in 2002, FDA established the Office of Combination Products (OCP) to address, among other issues, ambiguity on how to classify products that cross Center jurisdictions.[4] While OCP is not a review/approval Center for your combination product, they do provide general guidance on which pathway is best for your product and to ensure combination products have general oversight. Thus ultimately, if the sponsor cannot or does not want to decide, the Office of Combinations Products will decide the most important aspect of the combination, either the drug or the device, through the Request for Designation (RFD) process. If it’s deemed the drug is the primary mode of action, it will be reviewed by CDER (Center for Drug Evaluation and Research) and likewise, if its deemed the device is the primary mode of action, CDRH (Center for Devices and Radiological Health) will orchestrate the review.[5] It should be noted that the RFD process is currently undergoing changes to include the possibility of a pilot Pre-RFD.[6]
Are There Ever Cases Where Multiple Marketing Applications Are Required?
A single marketing application is sufficient for the review of most combination products. There are certain types of combination products where separate marketing applications for the individual constituent parts of the combination product will be reviewed by applicable centers, or there are times a sponsor sees merit in “choosing” this option. (e.g., new drug product exclusivity, orphan status, or proprietary data protection when two firms are involved).[7]
FDA has been responsive in accommodating tandem reviews. As recently as September 21, 2016, FDA released a draft guidance for the Coordinated Development of Antimicrobial Drugs and Antimicrobial Susceptibility Test Devices. The guidance is intended to “assist drug sponsors and device manufacturers who are planning to develop new antimicrobial drugs and AST devices and who seek to coordinate development of these products such that the AST device could be cleared either at the time of new drug approval or shortly thereafter.”[8]
What are Some Examples of FDA Combination Products?
Examples of combination products include pre-filled syringes, insulin injector pens, metered dose inhalers, transdermal patches, drug eluting stents, catheters with antimicrobial coatings, infusion pumps, medicated wound treatments and therapeutic orthopedic implants. In September 2015, Bayer Healthcare announced (FDA) approval of its at home use drug delivery device, BETACONNECT®, a first-of-its-kind electronic autoinjector indicated as a treatment for relapsing-remitting multiple sclerosis (RRMS) to deliver their drug Betaseron®. According to Bayer “all a patient will have to do is prepare the Betaseron® syringes provided in the medication pack and use BETACONNECT® for a more convenient and effective delivery”, that allows patient to select the speed and depth of injections as well.[9]
Testing your Drug Device Combination Product During Design Development
Whether your combination product is deemed a drug or medical device for regulatory purposes, testing for both may still be required. Device testing includes Human Factors Studies (or aka HF study). Representative users test the product to “help eliminate or mitigate any potential use-related hazards”, that may not be evident during the design planning. HF testing is usually done alongside or prior to clinical testing, however not all sponsors are aware of this requirement.[10] HF studies are generally conducted to evaluate user interfaces. It is not unusual for sponsors to ask FDA to clarify how HF concepts apply to the development of combination products.[11]
Because HF testing is still relatively new, inquiries to the FDA include:[12]
- What types of HF studies might need to be conducted for the combination product?
- When is the appropriate time to perform HF validation studies?
- What is the role of HF studies as compared to other types of clinical studies?
- Are additional HF studies necessary when the design of the combination product changes?
For the drug development side, expect consumer study requirements such as label comprehension (understanding the key label message), self-selection (choosing the right product), actual use (using according to labeled directions), or HF (interacting with the product).[13] Development of drug/device combination products are usually for convenience of the patient to self-administer, i.e. pre-filled syringes, insulin-injector pens. Therefore, there are many critical factors[14] to consider for patient safety that are not normally considered for patients in a clinical setting, i.e can they safely dispose of the product, can they safely self-administer, can they follow the included directions for administering? The objective is risk reduction by determining product user issues or related hazards with the products at the early design stage.
What’s next for FDA Combination Products?
The pipeline of combination products promises unprecedented drug delivery innovations. Integration of smart phone apps, global positioning technology and smart features built into devices and packaging are on the way. A quick search for innovative combination products found a tattoo-like skin patch for diabetes sufferers that can detect glucose levels in its wearer’s sweat and accordingly will deliver a drug as needed. While not approved as of this writing, innovations such as these are increasing in prevalence with growing consumer expectations related to ease of use, less restrictive living and integration with smart technologies.[15]
The earlier described BETACONNECT® will also feature an app to upload data to a smartphone or computer. The app is used to set reminder alerts for the next injection, with a calendar of all injections that have been scheduled, recorded, or missed. This injection history can be emailed directly to the patient or physician, so it is an added feature that benefits the patient and allows oversight by the healthcare provider.
As more products utilize sophisticated technology, FDA continues to respond in kind. In fact, the agency recently released a draft guidance entitled, “Software as a Medical Device (SaMD): Clinical Evaluation.” The draft guidance focuses on establishing the scientific validity, clinical performance, and analytical validity for these types of applications. The draft guidance was prepared via the support of the International Medical Device Regulators Forum (IMDRF), formerly the Global Harmonization Task Force (GHTF). IMDRF is a voluntary group of medical device regulators from around the globe, including FDA, who have come together to build on the strong foundational work of the GHTF. IMDRF has essentially consolidated forces with a focus on accelerating international medical device regulatory harmonization.
Drug-Device Combination Products are on the Rise
The drug and device industries recognize the demand for more convenience in our fast-paced lives and more advances in our tech savvy world, so they see value in investing money in new product design and development. Big pharma companies are hiring “their own device groups to develop devices specific to their therapies to set them apart in the marketplace”.[16]
- It is estimated that “approximately 1/3 of all medical products under development today are combination products and total sales could reach $115 billion dollars by 2019”.[17]
- “According to BCC Research, sales of the drug-device (segment of combination products alone) reached $21.4 billion in 2013 and $22 billion in 2014. This market is expected to grow to $31 billion in 2019, with a compound annual growth rate (CAGR) of 7.1% from 2014 to 2019.”[18]
- “The global drug device segment of the combination market accounted for USD 89.5 billion in 2015 and is anticipated to grow at a CAGR of over 7.9% during the forecast period.”[19]
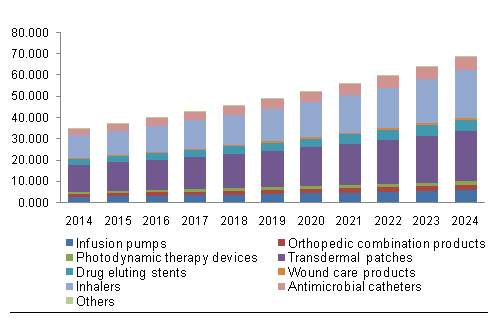
See below for a graph of current and expected North American sales for drug device combination products. It is also broken down by specific products. Note that transdermal patches lead the way.
North America Drug Device Combination Market, By Product, 2014 – 2024 (USD Billion)[20]
Figuring out the Regulatory Pathway for Combination Products
Regulatory expertise is critical to sponsors including determination of the best pathway for their specific product. It is important to have experts on both the drug and device side, not only for the purpose of initial regulatory strategy, but to ensure that through the entire product life cycle, (from design to manufacturing to market), the combination product meets or exceeds quality and regulatory requirements.
See PDG’s previous articles on FDA’s pre-sub program for how and why this program might benefit the developer of a combination product. Above all, if you need help with a pre-submission request, RFD (request for designation), or combination products generally, contact PDG! We have experts on staff with experience in drug, device and combination product registrations to assist you with all of your consultancy needs.
Jodi Hutchins is an Independent Regulatory and Quality Consultant with over 15 years of global medical device registration experience, to include FDA 510(k) submissions. She held her most recent position for 9 years, as QA/RA Director for a worldwide distributor of medical devices.
Charles Jaap is Vice-President of Operations and Business Development for PDG®, a global pharmaceutical and medical device consultant with extensive experience in the strategic development of drug products and medical devices.
The opinions and statements in this paper are solely those of Charles Jaap and Jodi Hutchins and do not necessarily reflect those of PDG©.





